
利用TBtools绘制简单的共线性图
学习目标:长颈鹿基因组文章中的基因组共线性关系图。
首先测试一下TBtools中在Graphics下面Advanced Circos工具。该工具下的三个输入框分别输入三个文件,文件中每列以制表符分隔开来。
①染色体的名字及其长度,共两列;
②染色体上所有元件特征的位置信息,分别为染色体名称、序列元件名称、序列原件的起始位置和序列原件的终止位置,共四列;
③同一元件在不同染色体的位置信息,分别为染色体1的名称、元件在染色体1的起始位置、染色体1的终止位置、染色体2的名称、元件在染色体2的起始位置、染色体2的终止位置。
为了方便测试,简单编辑了三个文件,格式如下
将这三个文件分别拖入输入框后,点击绘图,得到以下结果,表明没有软件报错,运行环境正常,如果用基因组大数据来绘图时,若文件格式正确,应该不会出问题。
接下来是如何在公共数据库中获取基因组数据,以及如何进行数据转换,得到circos工具需要的格式。以多孔鹿角珊瑚(Acropora millepora)基因组为例,该物种基因组是目前珊瑚虫纲为数不多的组装为染色体水平的基因组,包括已装配的14个染色体和为装配的部分。
...

词云图
利用wordcloud2包制作词云图,对数据的词频进行可视化。
准备excel表格,包括关键词和频数。例如:
word
freq
分子遗传学
9
分类学
9
生物地理学
8
古生物学
6
生态学
10
分子系统学
8
水生动物学
10
微生物
7
基因组学
8
地质学
5
气候学
6
环境海洋学
7
进化生物学
9
12345678setwd("D:/Desktop/词云图") #设置工作路径dir()install.packages("wordcloud2") #安装wordcloud2包library(wordcloud2)install.packages("openxlsx")library(openxlsx)wordmap<-read.xlsx("wordcloud.xlsx")wordcloud2(wordmap,size=0.3,shape='cardioid',color="random-l ...
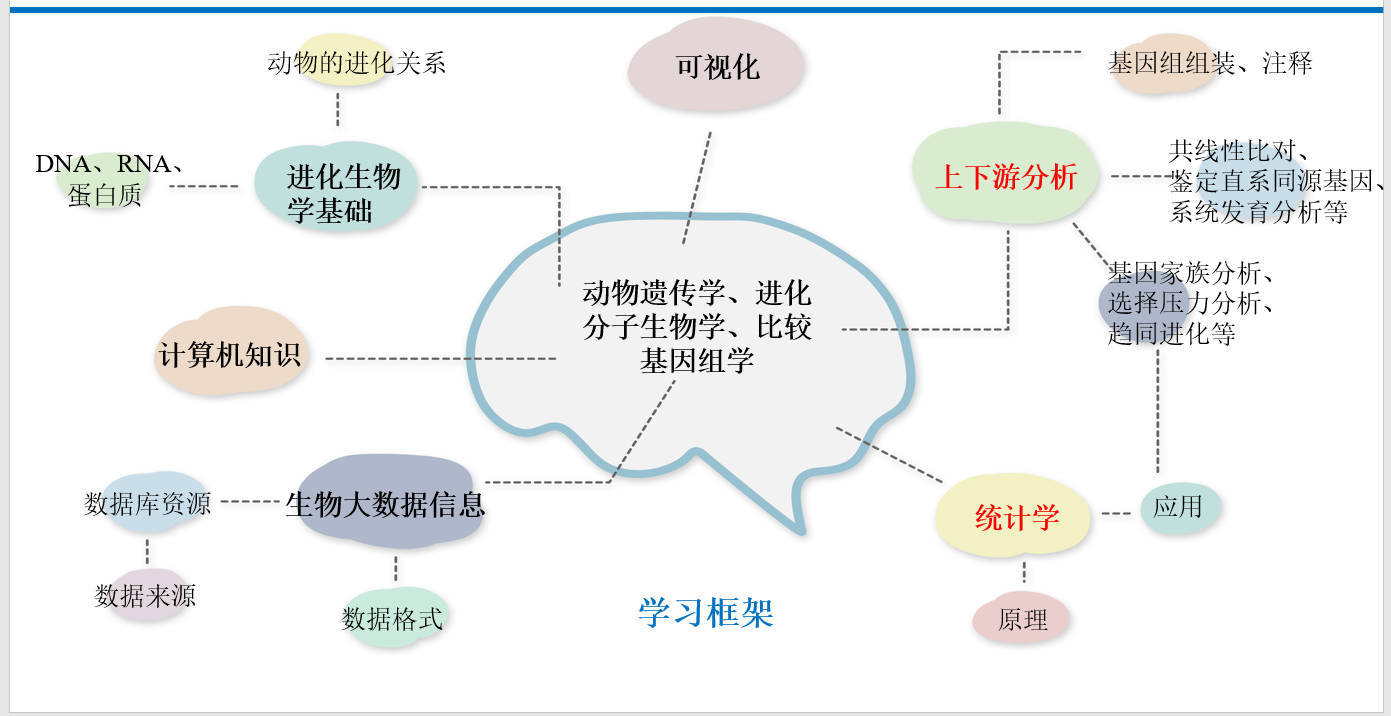
基因家族鉴定及分析
单物种基因家族鉴定及分析
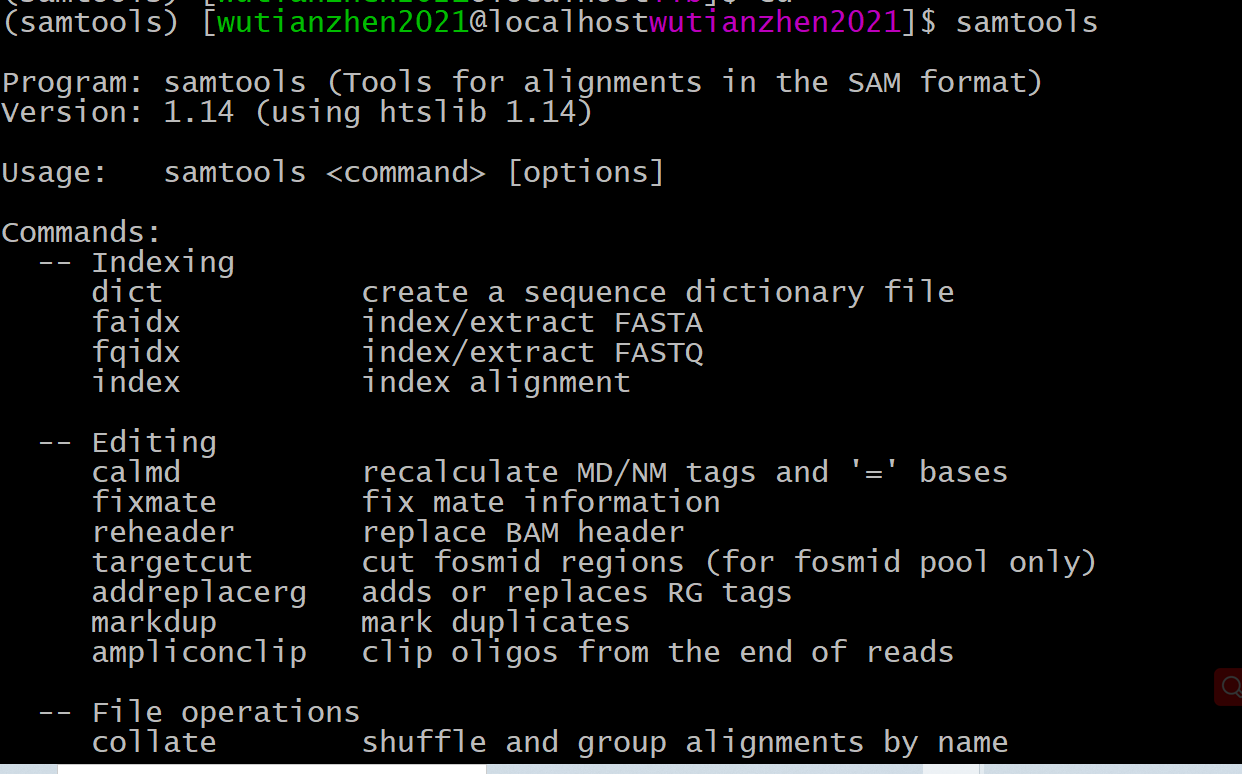
conda安装samtools时报错
当利用conda工具samtools后,如出现报错信息:
1error while loading shared libraries: libcrypto.so.1.0.0: cannot open shared object file: No such file or directory
解决方法如下
(1)在miniconda目录下samtools软件目录,进入lib文件查找libcrypto.so.*文件,对其进行软连接重命名至libcrypto.so.1.0.0。参考(14条消息) 解决samtools: error while loading shared libraries: libcrypto.so.1.0.0: cannot open shared object file_ET_April的博客-CSDN博客 和(14条消息) samtools: error while loading shared libraries: libcrypto.so.1.0.0: cannot open shared ……的解决方法_wyh0908的博客-CSDN博客
(2)有人说 ...
向国家生物信息中心数据库传输文件
利用Filezilla软件或Shell终端(FTP客户端)通过FTP协议向远程服务器(FTP服务器)进行大文件的传输
通过Linux中的ascp工具上传文件
1、通过Filezilla软件直接拖拽我是通过shell终端上传的,在Filezilla软件中的操作细节可能没遇到,比如如何设置二进制模式,若利用此途径可自行查阅相关帖子。
登录:输入地址、用户名和密码,然后快速链接
从左侧本地文件框中直接拖拽至右侧FTP远程服务器端位置,或者右键单击,点击上传即可
2、通过Shell终端传输(从下载ftp工具到传输文件)1234567891011121314yum -y install ftp #若ftp回车后显示没有安装,则可通过此命令进行安装ftp 服务器地址 #连接远程服务器Name: 用户名Password: 密码 #登录成功##以上传测序数据为例###ftp> cd GSA #同样地,利用cd命令切换路径250 Directory changed to /GSA ftp> binary #上传二进制模式文 ...
转录组测序研究进展
转录组(Transcriptome)是指特定细胞或组织中全部转录产物,包括信使RNA,核糖体RNA、转运RNA 以及非编码RNA。
1 转录组学是什么? 转录组学是从整体转录水平系统研究基因转录图谱并揭示复杂生物学通路和性状调控网络分子机制的学科。
2 转录组测序是什么? 转录组测序(RNA-seq)就是利用高通量测序技术将细胞或组织中全部或部分mRNA、small RNA和no-coding RNA 进行测序分析的技术
3 可以用来解决什么问题? ①检测与现有基因组序列相对应的转录本;②能发现和定量新的转录本;③基因转录的物种特异性和时空差异;④探究非编码RNA的调控机制;⑤单细胞转录组解析不同类型细胞的基因表达谱或空间分布情况。
怎么理解转录本与基因之间的关系呢?
研究一个基因时应该首先确定研究该基因的哪个转录本
转录本其实就是基因通过转录形成的一种或多种可供编码蛋白质的成熟的mRNA
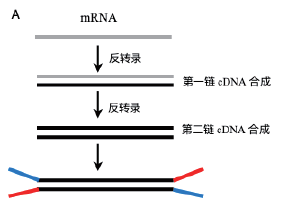
4 转录组测序的一般流程和测序内容?4.1 mRNA测序 利用mRNA 在3’ 端具有poly-A 的结构特 ...
test_pipeline-of-phylogeny
以2019年发表在MER期刊上的“Transcriptome-based target-enrichment baits for stony corals (Cnidaria: Anthozoa: Scleractinia)”文章中数据为测试数据,跑一遍构建物种分化时间树的探索流程如下:
1、数据获取在文献中获取452个直系同源基因序列文件,其中每个文件包括不同数量的物种,未进行多序列比对;分析所需软件和脚本:Mafft、Trimal、catfasta2phyml.pl、fasta2relaxedPhylip.pl、PartitionFinder、Iqtree、Raxml、astral.5.7.8.jar、Mcmctree等,多数可用conda安装,最好单独创建python2和python3环境用于以下软件的使用。
2、利用mafft软件进行多序列比对;123ls>temp.txt;sed -i 's/temp.txt//g' temp.txt;for i in `cat temp.txt`;do echo "mafft --maxiterate ...
batch sequences alignment using MAFFT
利用Mafft软件分别对蛋白和DNA进行批量比对
首先利用conda安装相关软件和脚本文件:
1234conda install mafft conda install Gblocksconda install trimalconda install pal2nal.pl
1、将DNA翻译成氨基酸进行多序列比对,再剪切,最后回译为DNA。将序列文件置于当前文件下 ./
1ls>temp.txt;sed -i 's/temp.txt//g' temp.txt;for i in `cat temp.txt`;do faTrans $i aa-$i;done;rm temp.txt;mkdir pepfile;mv aa-* pepfile #将DNA翻译成蛋白并移入pepfile文件夹;
1ls>temp.txt;sed -i 's/temp.txt//g' temp.txt;for i in `cat temp.txt`;do echo "mafft --maxiterate 1000 --localp ...
将fasta序列转化为键值对后进行序列处理
功能:将fasta格式的序列文件按照特定位置切分为前后两部分,并存为两个新的fasta文件。
其中,f是待切分的序列文件;f1、f2分别是切分后产生的文件;m为切割分界线位置;n为比对后序列全长。
关键知识为将fasta格式序列中的物种名定义为健、序列定义为值。
123456789101112131415161718192021222324f = open("PRM2-61.fas")f1=open("61-qian.fas","a")f2=open("61-hou.fas","a")m=144n=306seq = {}for line in f: if line.startswith('>'): name=line.replace('>','').split()[0] seq[name]='' else: seq[na ...



